
The incidence of obesity has been steadily increasing, tripling globally between 1975 and 2016, at a high cost to public health1. Obesity predisposes individuals to various diseases, including cancer, and the number of obesity-associated deaths globally each year1 (estimated at 2.8 million) is similar in scale to the reported COVID-19-associated deaths in the ongoing pandemic. Although fat-rich diets have taken much of the blame for the rise in obesity, excess consumption of processed sugars, and high-fructose corn syrup (HFCS) in particular, is strongly implicated in diet-induced obesity. Whether and how fructose causes obesity in humans remains a hotly debated question2,3. In a report in Nature that should make one think twice before gulping down sugar-sweetened drinks with fatty snacks, Taylor et al.4 propose that HFCS promotes obesity by boosting the ability of the intestine to absorb nutrients.
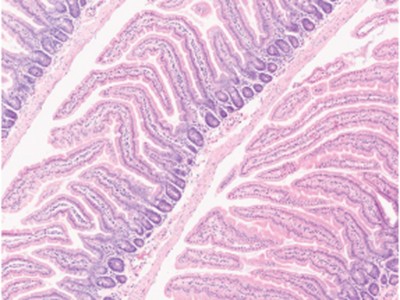
Read the paper: Dietary fructose improves intestinal cell survival and nutrient absorption
Evidence has emerged5–8 that the small intestine acts as the gatekeeper for the mammalian body against the harmful effects of fructose, the main one being the aberrant accumulation of fat (termed steatosis) in the liver. Moderate amounts of fructose — for example, those ingested when consuming fruits — are taken up and broken down by intestinal cells. Excess amounts, such as those that might be ingested after drinking a sugary beverage, overwhelm the intestine’s absorptive capacity and the fructose either ‘leaks’ into the bloodstream to reach the liver intact, or it spills over from the small intestine and reaches the colon5.
The breakdown of fructose in cells starts with its conversion to fructose 1-phosphate (F1-P). This modification involves the transfer of a phosphate group to fructose from the energy-providing molecule ATP, through the action of the enzyme ketohexokinase (KHK). Excess fructose in the liver fuels high KHK activity, which is thought to stimulate the expression of lipid-synthesis genes by diverse mechanisms9. The depletion of KHK in the liver of mice is enough to prevent fructose-induced liver steatosis6.
Fructose that ends up in the colon is broken down by resident bacteria to produce molecules that can then fuel lipid synthesis in the liver7. Furthermore, fructose increases intestinal ‘leakiness’, a condition in which loose connections between gut cells enable ingested nutrients, and toxins from bacteria in the colon, to escape to the liver, where they activate inflammatory signals from immune cells that boost steatosis8. Therefore, excess fructose harms the liver both directly and indirectly through changes in the intestine.
Taylor and colleagues’ study reveals that fructose has a previously unknown effect on the structure of the intestine (Fig. 1). Previous work10 had shown that HFCS promotes metabolic pathways that support the formation of colon tumours, so the authors wondered what consequences a HFCS-rich diet might have for non-cancerous intestinal cells. Taylor et al. found that HFCS-fed mice had longer intestinal protrusions — structures known as villi — and absorbed more dietary nutrients compared with mice that did not receive HFCS in their diet. Furthermore, fatty diets caused an even greater weight gain in mice if such diets also contained fructose than if they did not.

Figure 1 | Fructose-mediated gut changes. Nutrients in the gut enter the bloodstream after passing through cells in an intestinal protrusion called a villus. Cells at the tip of a villus have limited access to oxygen (a state called hypoxia), and they die for reasons such as energy depletion and oxidative stress. Taylor et al.4 report that, if mice received high-fructose corn syrup (HFCS) in their diet, they had longer villi and higher levels of lipids in their blood compared with animals that did not receive HFCS. The authors suggest that villus lengthening arises due to increased survival of hypoxic cells through changes triggered by the presence of fructose. Glucose from HFCS is metabolized, gaining two phosphate groups (red circles) to generate the molecule fructose 1,6-bisphosphate (F1,6-BP), which favours the formation of the high-activity, tetrameric version of the enzyme PKM2. This enzyme functions in a metabolic pathway called glycolysis. When the fructose from HFCS enters cells through the GLUT5 transporter, the enzyme ketohexokinase (KHK) converts it to fructose 1-phosphate (F1-P), which is structurally similar to F1,6-BP. In contrast to F1,6-BP, F1-P causes the dissociation of PKM2 subunits and shifts the balance from active PKM2 tetramers to low-activity PKM2 monomers. PKM2 monomers boost the survival of villus cells in hypoxia by suppressing oxidative stress and by inducing the expression of genes that help to sustain energy levels.
As a villus elongates when new cells are formed, the older intestinal cells move to the tip of the villus, away from blood vessels, and experience a gradually increasing limitation in oxygen availability (entering a state called hypoxia). The authors report evidence that at the tip of the villus, where hypoxia was extreme, cells were dying. Taylor and colleagues also found that new cells in the growing villi of HFCS-fed mice formed at similar rates to those of mice without HFCS in their diet. Together these observations suggest that hypoxia-induced cell death restricts villus length, and that the villi of HFCS-fed mice were longer because their cells were less likely to die compared with hypoxic cells in the villi of mice that were not fed HFCS.
Taylor et al. investigated how fructose might promote the survival of hypoxic cells. The authors’ analysis of cells grown in vitro revealed that fructose drove an increase in the level of F1-P. Moreover, treatment of these cells with fructose inhibited the metabolic pathway of glucose breakdown called glycolysis at the step catalysed by the enzyme PKM2. Low PKM2 activity helps to reroute intermediate molecules from the glycolysis pathway to biosynthetic and antioxidant pathways that enable cells to proliferate and survive under stress11. Various intracellular molecules bind to and switch PKM2 between a high-activity tetrameric form (containing four protein subunits) and a low-activity monomeric form (containing one protein subunit), depending on the state of the cell11. Monomeric PKM2 can move to the nucleus to support the transcription factor HIF-1α in driving the expression of genes that help hypoxic cells to maintain their energy reserves12.
Deciphering metabolism, one microbe at a time
The authors found that F1-P binds to PKM2, probably by virtue of its structural similarity to fructose 1,6-bisphosphate (F1,6-BP), a molecule of the glycolysis pathway that can activate PKM2. In contrast to F1,6-BP, F1-P drives PKM2 to adopt a low-activity monomeric form. This helps fructose-fed hypoxic intestinal cells to lessen oxidative stress and boosts HIF-1α activity to preserve energy levels. When Taylor and colleagues gave mice TEPP-46, a small molecule that locks PKM2 into an active tetrameric form that evades inhibition by F1-P, this prevented villus elongation in HFCS-fed mice, indicating that fructose-derived F1-P helps hypoxic cells in the growing villus to survive by controlling PKM2.
To further investigate the role of PKM2 and KHK in villus elongation, the authors deleted either the part of the Pkm gene that specifically encodes PKM2 or the Khk gene. The modification of Pkm to prevent PKM2 expression drove the expression of an alternative version of Pkm that encodes a protein called PKM1, which is active all the time11, similarly to TEPP-46-bound PKM2. These genetic deletions prevented villus elongation as a consequence of a HFCS diet and, importantly, reduced the level of blood lipids in HFCS-fed mice compared with the level in mice in which the genes were not deleted. This suggests that fructose-induced villus lengthening enables greater absorption of dietary fat.
Taylor et al. then investigated whether their findings in the normal intestine were also relevant to intestinal tumours. They found hypoxic cells in intestinal tumours in mice that were genetically predisposed to develop these tumours. As previously described10, feeding such mice HFCS led to larger tumours compared with feeding them diets without HFCS. Although treatment of HFCS-fed mice with TEPP-46 did not eliminate these tumours, it prevented the tumour-promoting effects of HFCS. The authors also observed increased expression of HIF-1α target genes and low PKM2 activity in samples of human colorectal tumours compared with nearby, non-cancerous, tissue. Together, these findings suggest that — similarly to the situation for hypoxic cells in growing villi — a low-activity state of PKM2 could also support tumour growth by enabling intestinal cancer cells to survive hypoxia.
Designer fibre meals sway human gut microbes
This work provides compelling evidence that PKM2 and its newly described ligand F1-P mediate previously unappreciated effects of fructose in both normal and cancerous intestinal tissues. The findings also raise several questions relating to our current understanding of how the intestine handles nutrients. In healthy animals, gut enzymes called lipases break down ingested lipids into products that are absorbed by intestinal cells and repackaged for delivery through the lymphatic system and into the blood. So the question of how ingested dietary lipids cause a rise in lipids in the blood, as they seem to do in HFCS-fed mice even when lipases are inhibited, according to Taylor and colleagues, begs further investigation. Perhaps villus elongation is linked to intestinal leakiness8 and, when fatty foods overwhelm the enzymatic capacity of lipases, excess dietary lipids bypass intestinal cells and leach into the bloodstream.
Fructose consumption increases the level of expression of the GLUT5 transporter that aids fructose entry into cells5, thereby enhancing fructose uptake. It is unclear whether villus elongation provides an additional boost to fructose uptake, which might decrease fructose spillover into the colon and the liver as a consequence of persistent fructose consumption5. In such a scenario, even though villus elongation promotes fat absorption, it might also partly mitigate the harmful effects of fructose itself on the liver. It will be interesting to determine how the complex balance between the intestine’s absorptive and metabolic capacities for fructose, lipids and other nutrients, and the intersection of these processes with the activity of microorganisms in the colon together contribute to obesity-related conditions.
Considering the therapeutic potential of this work, the authors’ experiments strongly suggest that PKM2 supports the survival of the intestinal cells in which it is expressed. However, PKM2 is also expressed in many other types of cell, such as immune cells, which mediate the functions necessary to maintain the health of both the intestine and the liver.
Curiously, despite its ability to prevent villus elongation mediated by HFCS, TEPP-46 did not improve liver steatosis in the authors’ experiments. This finding contrasts with the decrease in fructose-induced steatosis that the authors observed with the Pkm deletion that prevents PKM2 expression. This discrepancy between genetic and pharmacological approaches to PKM2 modulation raises the possibility that the combined action of PKM2 activators on multiple cell types might ultimately determine their ability to modulate specific tissue functions in disease. Despite early hopes, PKM2 activators have yet to reach the clinic as cancer therapies. Taylor and colleagues’ work highlights the fact that gaining an in-depth understanding of tissue and disease contexts could allow researchers to suggest new therapeutic areas in which PKM2 activation might prove useful. Regardless, avoiding sugary drinks altogether might be a good start to curbing obesity.
doi: https://doi.org/10.1038/d41586-021-02195-1
References
- 1.
Lancet Gastroenterol. Hepatol. 6, 411 (2021).
- 2.
van Buul, V. J., Tappy, L. & Brouns, F. J. Nutr. Res. Rev. 27, 119–130 (2014).
- 3.
Stanhope, K. L. Crit. Rev. Clin. Lab. Sci. 53, 52–67 (2016).
- 4.
Taylor, S. R. et al. Nature https://doi.org/10.1038/s41586-021-03827-2 (2021).
- 5.
Jang, C. et al. Cell Metab. 27, 351–361 (2018).
- 6.
Andres-Hernando, A. et al. Cell Metab. 32, 117–127 (2020).
- 7.
Zhao, S. et al. Nature 579, 586–591 (2020).
- 8.
Todoric, J. et al. Nature Metab. 2, 1034–1045 (2020).
- 9.
Softic, S., Cohen, D. E. & Kahn, C. R. Dig. Dis. Sci. 61, 1282–1293 (2016).
- 10.
Goncalves, M. D. et al. Science 363, 1345–1349 (2019).
- 11.
Dayton, T. L., Jacks, T. & Vander Heiden, M. G. EMBO Rep. 17, 1721–1730 (2016).
- 12.
Luo, W. et al. Cell 145, 732–744 (2011).